
La Esclorosis Lateral Amiotrófica (ELA), también llamada enfermedad de Lou Gehrig, es una enfermedad neurodegenerativa que destruye las neuronas motoras, importantes para todas las funciones vitales, incluida la respiración.
Se trata de una enfermedad mucho más frecuente en personas mayores, pero que también puede afectar a personas de entre 20 y 30 años. Su prevalencia es de aproximadamente dos porcada 100.000 personas y la causa de la enfermedad no siempre es la misma, aunque hay una serie de factores genéticos que influyen en el inicio de la misma.
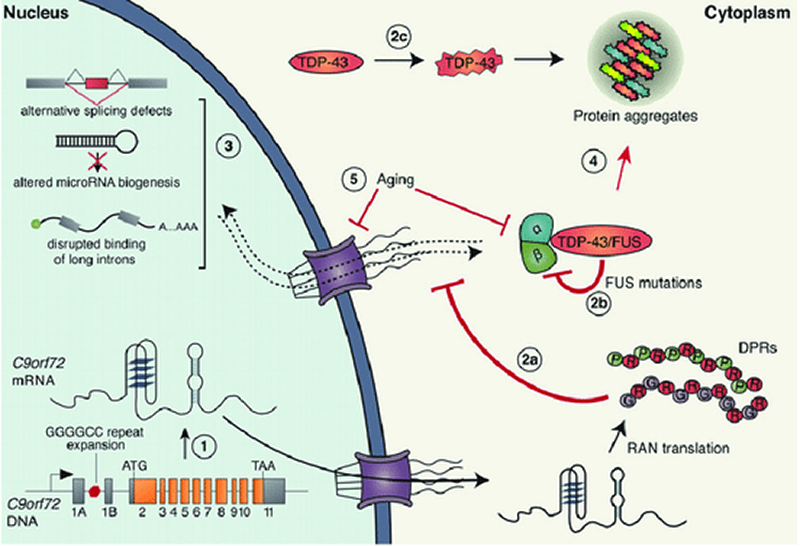
En la ELA una cadena de ADN se repite de forma errónea, concretamente la expansión repetida de nucleótidos (GGGGCC) en el primer intrón del gen C9orf72 parece ser la causa más común tanto de ELA como de Demencia Frontotemporal (DFT).
Esta expansión de nucleótidos genera unos agregados proteicos que se acumulan en el cerebro de los pacientes, interfiriendo con las neuronas sanas y bloqueando la capacidad de las células para funcionar normalmente.
Hay también otros genes asociados con el desarrollo de la enfermedad y agregados celulares. Algunos de los más conocidos son: SOD1, TARDBP, FUS, TDP-43, OPTN y UBQLN2, aunque existen muchos más.
Índice de contenidos
Gen RPS25
Recientemente, investigadores de la Facultad de Medicina de la Universidad de Standford, en Estados Unidos, han publicado un artículo en la revista Nature Neuroscience donde han identificado un gen, RPS25 (Small Ribosomal Protein Subunit 25) que codifica una proteína ribosomal y podría convertirse en una buena diana terapéutica para la ELA, DFT y otras enfermedades neurodegenerativas ligadas a la expansión de repeticiones de nucleótidos.
“Sabemos que estos agregados ricos en proteínas son un sello distintivo claro de la ELA. Pero este hallazgo nos permite un análisis más profundo de cómo se hacen esos agregados y, potencialmente, cómo podemos obstaculizar ese proceso”. Explica el profesor de genética Aaron Gitler en un artículo en Nature Neuroscience.
Cuando la actividad del gen RPS25 se elimina se observa que los niveles de proteína descienden en aproximadamente un 50% en todos los ámbitos.
Traducción RAN
Durante la formación de proteínas en la célula, el ribosoma que reside en la célula procesa el ARN mensajero (ARNm) que está basado en ADN y lo convierte en proteína. Este proceso denominado traducción se inicia mediante un código en el ARNm que indica al ribosoma donde comenzar a traducir (triplete AUG).
Lo que ocurre con las repeticiones del ADN asociadas a ELA es que no tienen este código de inicio a diferencia del ARNm normal y, como indica el autor del artículo, “la traducción regular no funciona con las repeticiones”.
No obstante, existe una solución molecular alternativa a este proceso. Se trata de una traducción no convencional llamada traducción no AUG asociada a repetición o traducción RAN que es la causante de la generación de unos dipéptidos (DPR). Éstos son propensos a agregarse entre ellos y su acumulación en el sistema nervioso central es la que provoca estas enfermedades.
El mecanismo exacto de la traducción RAN no está del todo claro, pero tal como se demuestra en este artículo, estudios hechos tanto en levaduras como en una línea celular humana (Hap1) demuestran la disminución de la traducción de ARN GGGGCC en células donde se ha eliminado la expresión del gen, RPS25 KO.
Estos datos confirman la existencia de un papel importante del RPS25 en la traducción RAN de la expansión C9orf72.
Para probar si la inhibición de RPS25 mitigaba fenotipos neurodegenerativos causados por repeticiones en el C9orf72 in vivo, se utilizó Drosophila transgénica con una expansión de 36 repeticiones y neuronas humanas de pacientes con ELA. Se vio cómo en los dos casos se produce una reducción de los productos de la traducción RAN y una consiguiente mejora en la esperanza de vida.
Otras enfermedades raras
El equipo también ha probado la función del RPS25 en células humanas que modelan la enfermedad de Huntington y la ataxia espinocerebelosa, SCA2, otras dos enfermedades neurodegenerativas que tienen características de agregados similares a la ELA. Así lo explica Yamada en su artículo y también allí se ha visto que la inhibición del gen ayudó a reducir los niveles de proteínas dañina.
Futuros avances
Todavía es una etapa preliminar pero obstaculizar el gen RPS25 parece ser un objetivo prometedor para reducir las proteínas destructivas observadas en ELA e incluso alargar su vida útil, como explica Yamada.
“Los hallazgos son intrigantes, pero antes de que los científicos puedan comenzar a buscar RPS25 como un objetivo de fármaco, el equipo tiene que dar más pasos”, admite Yamada en su artículo.
Ahora están investigando cómo un modelo animal más complejo como un ratón sería adecuado sin RPS25. «Con las moscas de la fruta, manipulamos el gen; no lo eliminamos por completo”, apunta Yamada.
“Si un animal puede sobrevivir sin el gen por completo es una parte importante de nuestro próximo paso». Además, siguen buscando una imagen más clara de la traducción de RAN en humanos en general. «¿Ocurre solo en condiciones neurogenerativas? ¿O hay un papel más amplio para eso en individuos sanos? Todavía no sabemos la respuesta a esas preguntas y será crucial resolverlo antes de perseguir al RPS25 como un objetivo terapéutico». Se cuestionan.
Todavía es pronto, pero está claro que estamos ante una nueva proteína involucrada en traducción RAN y que sugiere estrategias para inhibir la función de RPS25, que podrían perseguirse como una terapia para ELA, DFT y otras enfermedades neurodegenerativas causadas por repetidas expansiones de nucleótidos.