
La Atrofia Muscular Espinal (AME) es una enfermedad neuromuscular autosómica recesiva causada por la pérdida de función del gen de supervivencia de la neurona motora (SMN1), que se localiza en el cromosoma 5. Tiene una incidencia aproximada de 1/10000 nacimientos y una frecuencia de portadores alrededor de 1/50. La enfermedad se caracteriza por la degeneración de las motoneuronas alfa del asta anterior de la médula espinal, lo que produce una debilidad simétrica y atrofia progresiva de los grupos musculares proximales.
La Atrofia Muscular Espinal es considerada como una enfermedad rara, pero es una de las principales causas hereditarias de mortalidad infantil.
Según la gravedad de las manifestaciones clínicas, la edad de aparición y la evolución de las mismas se clasifica en tres grupos (tipo I o aguda, tipo II o intermedia y tipo III o crónica), que guardan una estrecha relación con el número de copias de un gen homólogo a SMN1 conocido como SMN2.
En realidad, estos genes son dos copias invertidas separadas por unas 500 Kb en el locus SMN localizado en el brazo largo del cromosoma 5 (5q13), siendo SMN1 la versión telomérica determinante de la Atrofia Muscular Espinal y SMN2 la versión centromérica asociada al grado de severidad de dicha enfermedad (Prior et al. 2011). Estos dos genes tienen promotores equivalentes y se diferencian únicamente en 5 nucleótidos en su extremo 3’ (en el intrón 6, exón 7, intrón 7 y exón 8).
La diferencia principal se encuentra en un nucleótido del exón 7 (840C>T), que altera el splicing del RNA y hace que generalmente el mRNA de SMN2 carezca del exón 7, resultando en una reducción significativa (80-90%) de los niveles de proteína SMN funcional (Stabley et al. 2015; Tisdale & Pellizzoni, 2015). Por otro lado, el gen SMN1 genera el transcrito completo incluyendo el exón 7.
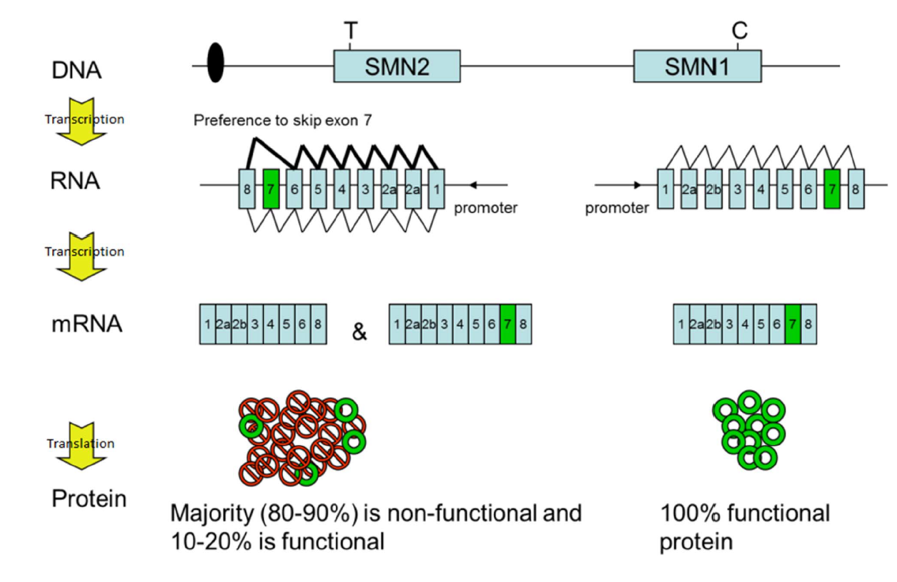
La patología molecular de aproximadamente el 95% de los casos de Atrofia Muscular Espinalconsiste en la ausencia homocigota del exón 7 de SMN1 debido a deleciones o conversiones génicas. También se han descrito mutaciones menos frecuentes que afectan a algunos nucleótidos del gen (Wirth et al. 2000).
En consecuencia, los pacientes con Atrofia Muscular Espinal producen cantidades insuficientes de proteína SMN de longitud completa o proteínas no funcionales e inestables (Tizzano, 2007; Prior et al. 2011). Este hecho se ve compensado por la pequeña cantidad (alrededor de un 10%) de proteína SMN funcional traducida a partir del gen SMN2, sin embargo, esta compensación no es completa y no evita el desarrollo de la enfermedad.
Aun así, SMN2 tiene un papel muy importante influyendo en la gravedad de la atrofia (Tisdale & Pellizzoni, 2015) y se ha demostrado en diversos estudios que, por regla general, a mayor número de copias del gen el fenotipo es menos grave. Los pacientes con AME tipo I son generalmente los que han sufrido deleción de SMN1 y suelen tener una o dos copias del gen SMN2, mientras que los pacientes con tipo II y III muestran con mayor frecuencia conversión del gen SMN1 a SMN2 y normalmente tienen de 3 a 5 copias de SMN2 (Velasco et al. 1996; Lefebvre et al. 1995; Cuscó et al. 2001). Se ha llegado a describir casos asintomáticos con deleción homocigota de SMN1 y con 5 copias de SMN2 (Prior et al. 2004).
> Dado que la mayoría de pacientes de Atrofia Muscular Espinal presentan deleción homocigota del exón 7 de SMN1, el análisis genético de esta región cromosómica es el método de elección para la confirmación del diagnóstico. Los métodos moleculares empleados permiten determinar el número de copias de SMN1 y detectar portadores de la enfermedad, siendo las técnicas de MLPA (multiplex ligation-dependent probe amplification) y PCR cuantitativa las más utilizadas actualmente. Además, hay laboratorios que usan el test RFLP (restriction fragment length polymorphism) para un análisis cualitativo de la deleción y la secuenciación para la identificación de las formas heterocigotas que presentan deleción en un cromosoma y otro tipo de mutación en el cromosoma homólogo.
> El análisis del número de copias de SMN2 no se realiza de manera rutinaria para el diagnóstico o para test de portadores de Atrofia Muscular Espinal, sin embargo puede ser muy útil en ensayos clínicos y screening de recién nacidos con el fin de identificar pacientes con mayor probabilidad de respuesta a estrategias terapéuticas basadas en la activación de la expresión de proteína SMN completa a partir de SMN2. En el contexto del diagnóstico clínico y prenatal el estudio del número de copias de SMN2 debe ser interpretado con cautela, ya que como sabemos existen variantes de la secuencia de este gen y otros genes que también pueden modificar el fenotipo de la enfermedad. Además, tanto para SMN1 como para SMN2, los métodos actuales de cuantificación del número de copias no permiten determinar si estas copias se encuentran en cis (en un único cromosoma) o en trans (en ambos cromosomas), lo que hace más difícil el consejo genético para los casos portadores (Prior et al. 2011).
En cualquier caso, los métodos cuantitativos mencionados también incluyen la detección de SMN2 de forma simultánea a la de SMN1. La mayoría de laboratorios dedicados a estas pruebas utilizan el test MLPA por ser un método muy preciso y sensible, sin embargo, el protocolo de trabajo es largo y el análisis puede resultar en ocasiones complicado. Una alternativa mucho más sencilla y rápida es el kit Amplidex PCR/CE SMN1/2 que acaba de lanzar al mercado la compañía Asuragen, con la que trabajamos en Rafer desde hace 9 años. El kit Amplidex PCR/CE SMN1/2 es un ensayo multiplex de alta especificidad con el que se amplifica el exón 7 de los genes SMN1 y SMN2 y un control endógeno (EC) de DNA genómico purificado. Los amplicones marcados con fluorescencia se analizan por electroforesis capilar, de forma que podemos distinguir por el tamaño en pares de bases los fragmentos correspondientes al control endógeno y a los genes SMN1 y SMN2. Además, los primers están diseñados para detectar las conversiones entre ambos genes como picos únicos en el electroferograma. Estos picos se denominan híbridos, ya que representan fragmentos con secuencias de los dos genes: el pico híbrido SMN1 indica conversión del gen SMN2 a SMN1 y tiene secuencia de SMN1 en el exón 7 y secuencia de SMN2 en el intrón 7, y el pico híbrido SMN2 indica conversión de SMN1 a SMN2, con secuencia de SMN2 en el exón 7 y secuencia de SMN1 en el intrón 7.
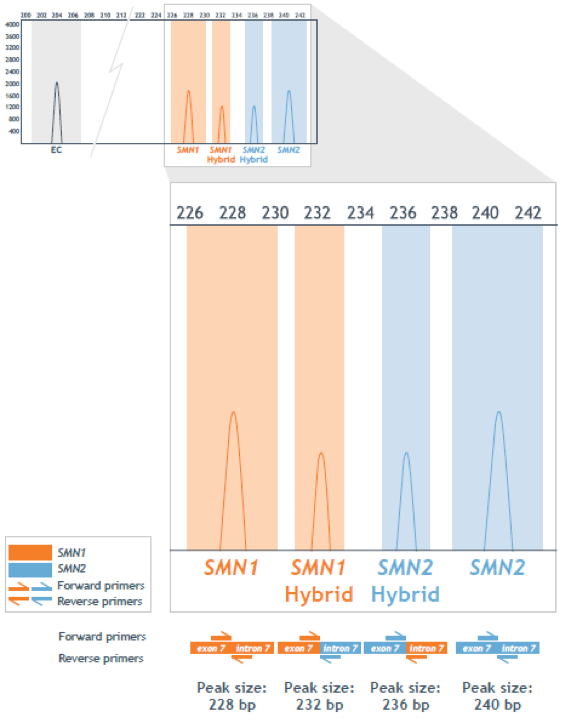
Para determinar el número de copias del exón 7 en ambos genes se analiza el área bajo la curva del pico correspondiente y se normaliza con el área del pico del control endógeno. El resultado final es la suma de las copias presentes en los fragmentos específicos de cada gen y las copias de los fragmentos híbridos.Así, con este kit podemos diferenciar de forma precisa 0, 1, 2, 3 o ≥ 4 copias de ambos genes.
En comparación con el método MLPA, el kit Amplidex reduce el tiempo de trabajo manual a casi la mitad y el tiempo de procesamiento automático es aproximadamente 10 veces menor. Además, el análisis es más sencillo gracias a una herramienta macro que calcula los ratios normalizados de las áreas de los picos y los convierte en número de copias de forma automática, indicando también en el informe la presencia de picos híbridos resultantes de la conversión génica.
Recientemente se ha completado la evaluación de este kit en varios centros de referencia europeos y en España se están realizando las primeras pruebas en el laboratorio de Genética del Hospital Vall d’Hebron de Barcelona, bajo la responsabilidad del Dr. Eduardo Tizzano, investigador referente de la Atrofia Muscular Espinal. Estos primeros ensayos arrojan resultados prometedores de gran valor para la confirmación del diagnóstico y el asesoramiento genético y también para la aplicación de una terapia eficaz. A partir de ahora nuestra labor desde Rafer es promocionar y ayudar en la puesta a punto de este test en los hospitales de referencia en el diagnóstico de Atrofia Muscular Espinal en España y Portugal.
Si quieres saber más puedes solicitar información
Artículos relacionados
Efectos de un bajo número de repeticiones CGG en el gen FMR1